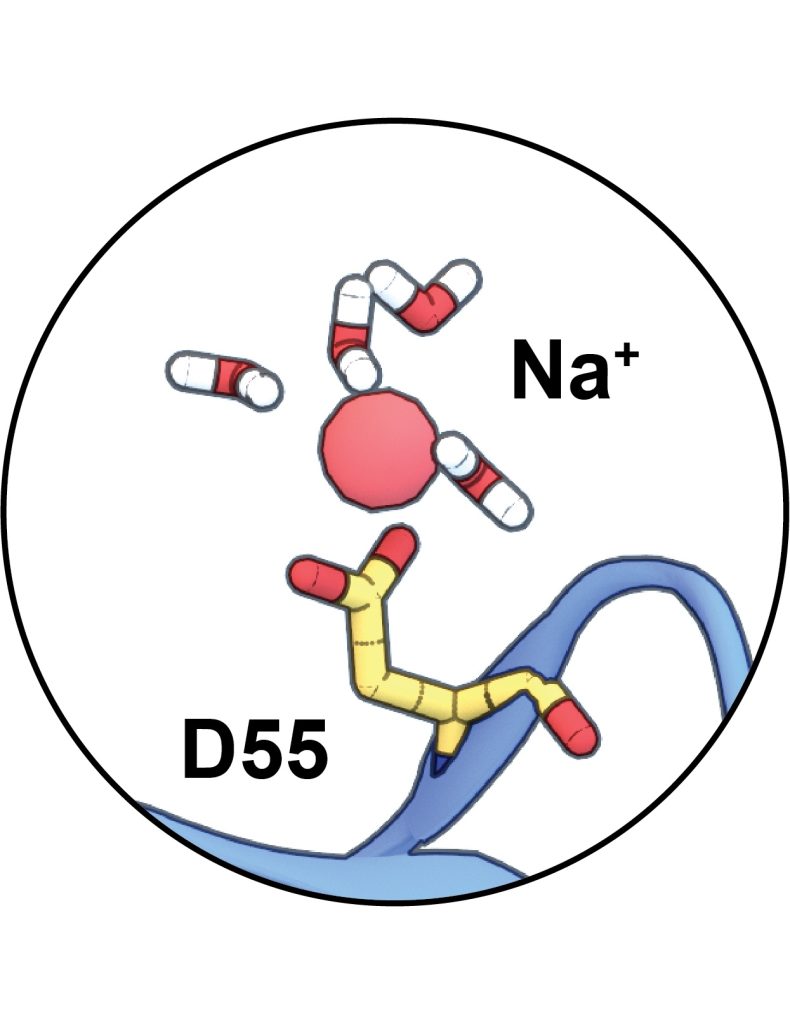
Our current research focuses on the parameterization of novel ligands, particularly in relation to their binding properties with claudin-15. We explore a range of ligands using various computational approaches, including molecular dynamics (MD) simulations and docking, applied across diverse datasets. This allows us to evaluate both the structural and functional compatibility of new ligands. Depending on the goal of the machine learning model, we can train it using our dataset along with the corresponding docking information to improve predictions of ligand–target interactions.